
All-in-one guide to Understanding the Chinese Drug Master File (cDMF)
Learn everything about cDMF, and prepare your entry into the Chinese regulation market
- Introduction
- The Role of the FDA for the Drug Master File (DMF)
- What is a Drug Master File (DMF)?
- Chinese DMF: What is it?
- Why Your Company Needs a Chinese DMF
- The Path to cDMF
- Key Components of a China Drug Master File
- What Makes the cDMF So Challenging?
- Finding Expert cDMF Guidance
- Conclusion
Introduction
Hello Pharma enthusiasts, Pharmaoffer here with a brand new blog filled with pharmaceutical insights, designed to help you navigate your company in the intricate world of pharmaceuticals.
Today, we are diving deep into the Chinese Drug Master File (cDMF), which you may have heard about if you have ever registered a foreign product to enter the Chinese market. We are going to discuss everything about cDMF and more, starting with the basics, like the role of the FDA, what a Drug Master File (DMF) really is, and how it works.
If you are here to find assistance in submitting your Chinese DMF, we’ll give you permission to skip right over to the later section of the blog, where we will introduce Accestra, Pharmaoffer’s new partner for helping companies enter the Chinese market easily and without delays or setbacks.
Whether you’re an API supplier, a manufacturer, or part of the pharmaceutical production chain, this blog is packed with essential tips and guides to help you succeed in the Chinese market.
Let’s get started!
The Role of the FDA for the Drug master file (DMF)
To understand the DMF, it’s helpful to know the institution’s role that impacts it most, namely the Food and Drug Administration, better known as the FDA.
The FDA, the federal agency under the United States Department of Health and Human Services, plays a crucial role in the pharmaceutical industry. Its primary responsibility, as stated on its website, is to safeguard public health. It does this by ensuring the safety, efficacy, and security of human and veterinary drugs, biological products, medical devices, food supply, cosmetics, and products that may emit radiation. Additionally, the FDA regulates the manufacturing, marketing, and distribution of tobacco products, an authority granted to the FDA under the Family Smoking Prevention and Tobacco Control Act (Tobacco Control Act) signed into law in 2009.
Because of the role and importance of the US market in terms of manufacturing and innovation (the USA is the top pharma innovation country in the world, according to statists), FDA approval is often considered a gold standard in the pharmaceutical industry. This is something all new products everywhere in the world aim to have because drugs and medical devices approved by the FDA are easily accepted in international markets, facilitating global trade and access to innovative treatments without compromising worldwide health.
What is a Drug Master File (DMF)?
The FDA created the DMF concept to facilitate pharmaceutical manufacturers’ submission of confidential information. This is the primary purpose of a DMF: allowing the FDA to review detailed proprietary information about drug substances and components without requiring the information to be included in a public drug application. The benefits of DMFs are:
- Facilitating Drug Approvals: DMFs allow multiple drug applications (NDAs, ANDAs) to reference the same confidential information without duplicating the submission, streamlining the drug approval process.
- Ensuring Uncompromised Quality and Safety: The FDA relies on the information in DMFs to meticulously verify that drug substances and components adhere to the stringent standards for quality, safety, and efficacy. This proves the crucial role of DMFs in the regulatory process.
Chinese DMF: what is it?
A China Drug Master File (cDMF), is analogous to the Drug Master File used in the United States but is specific to the regulatory requirements of China, the second-largest market in the world.
The cDMF is part of China’s regulatory framework for pharmaceutical products, managed by the National Medical Products Administration (NMPA), formerly known as the China Food and Drug Administration (CFDA).
Why Your Company Needs a Chinese DMF
If you are looking to expand your pharmaceutical endeavors into Chinese territories, there is no way around a China DMF. The regulatory demands within China’s pharmaceutical and medical device sector, particularly for API suppliers and manufacturers, are undeniably intricate to navigate. If you are an outsider, it’s even harder because of the language and culture barriers you may face along the way. Let’s overview the steps for your pharmaceutical company in the next section.
Want to be sure you’re paying a fair price for your APIs? Now it’s easy.
Pharmaoffer’s Trade Data service gives you an overview of the latest average selling prices, transaction volumes, and tracking search data across the globe for thousands of APIs.
Simplify your sourcing process by accessing current market insights and detailed transaction histories that will give your business a competitive edge.
Check it out today and make smarter sourcing decisions!
The Path to cDMF
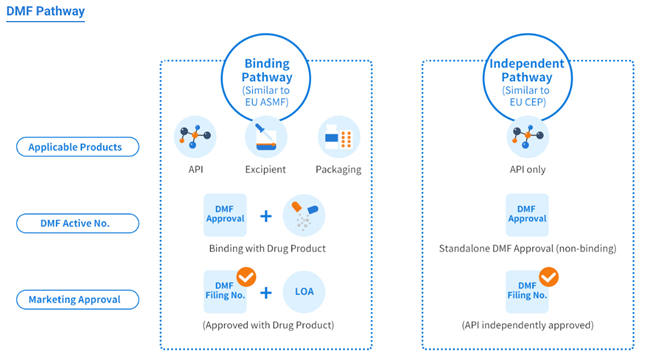
Like the U.S. DMF, the China DMF encompasses three product scopes/categories and provides two registration pathways that cater to different components of pharmaceutical manufacturing:
Product categories:
- Active pharmaceutical ingredients (APIs)
- Excipients
- Packaging materials
Registration Pathways:
- Binding review pathway
(similar to the EU ASMF, covering all three product categories).
This pathway is similar to the European Union’s Active Substance Master File (EU ASMF) system and it is applicable to all three product categories: APIs, excipients, and packaging materials. The Binding Review pathway ensures that all aspects of the drug product, including its components, are thoroughly evaluated together. - Stand-alone review pathway
(similar to the EU CEP, applicable to API products only)
The Stand-alone review pathway is similar to the European Union’s Certificate of Suitability (EU CEP) system and it is specifically applicable only to APIs, which is the main difference between the Bind Review Pathway. Stand-alone streamlines the approval process for APIs, allowing manufacturers to gain approval for the API once and reference it in multiple drug applications, saving time and resources. As we can see in the image below.
Key Components of a China Drug Master File
In previous sections, we discussed that the cDMF is a detailed, comprehensive document that includes information on the manufacturing, processing, packaging and storage of drug products. In this section, we discuss what paperwork the China DMF actually includes, or the key components that ensure NMPA has all the necessary information to evaluate the quality, safety, and efficacy of the drug. So you have a better understanding of just how comprehensive it is. Here are the primary sections and their contents:
1. Administrative Information:
Cover Letter: A summary of the cDMF submission, including purpose and scope.
Table of Contents: An organized list of all sections and subsections within the cDMF.
Applicant Information: Details of the company submitting the cDMF, which includes contact information and the local legal entity acting as the registration representative.
2. Drug Substance Information:
General Information: Includes the name, structure, and properties of the drug substance.
Manufacturer Details: Information about the manufacturing site, including addresses, contact information, and any relevant certifications or licenses.
Manufacturing Process and Controls: A detailed description of the manufacturing process, including the steps involved, in-process controls, and critical parameters. This section also includes batch records and flow charts.
Control of Materials: Information on the raw materials, reagents, and solvents used in the manufacturing process, including their sources and specifications.
Characterization: Data on the chemical structure, purity, and other characteristics of the drug substance.
Specifications and Test Methods: Detailed specifications for the drug substance and the analytical methods used to ensure compliance with these specifications.
Stability Data: Stability studies and data supporting the shelf life and storage conditions of the drug substance.
3. Drug Product Information (if applicable):
Formulation Details: Information about the composition of the drug product, including active and inactive ingredients.
Manufacturing Process: Description of the manufacturing process for the drug product, including the steps involved, in-process controls, and critical parameters.
Packaging Information: Details about the packaging materials and conditions used for the drug product, including primary and secondary packaging.
Specifications and Test Methods: Specifications for the drug product and the analytical methods used to ensure compliance with these specifications.
Stability Data: Stability studies and data supporting the shelf life and storage conditions of the drug product.
4. Facilities and Equipment:
Facility Descriptions: Detailed descriptions of the manufacturing and testing facilities, including layouts, equipment, and environmental controls.
Equipment Information: Information about the equipment used in the manufacturing and testing processes, including qualifications and maintenance records.
5. Quality Control:
Quality Assurance: Overview of the quality assurance systems in place, including standard operating procedures (SOPs), quality manuals, and training programs.
Release Testing: Procedures and criteria for the release of the drug substance or product, including sampling methods and acceptance criteria.
Stability Testing: Protocols and results for stability testing, including conditions, durations, and testing intervals.
6. Environmental Impact:
Environmental Assessments: Information on the potential environmental impact of the manufacturing process, including waste management and disposal methods.
Risk Management Plans: Plans to mitigate environmental risks associated with the manufacturing process.
What makes the cDMF so challenging?
It’s easy to understand that with such extensive and complex information, as we’ve seen in the previous section, the China DMF can be challenging to get. But that’s not all you have to deal with in order to get cDMF. The compliance landscape is constantly evolving often leading to delays and even rejections of product registrations for companies, which can be very frustrating. Lastly, and despite aligning with international regulations like the ICH (International Council for Harmonisation of Technical Requirements of Pharmaceuticals for Human Use), China still requires a local legal entity to act as the registration representative for non-Chinese companies.
Needless to say if your company fails to find a local representative with expertise in China’s Regulatory Affairs it will, most certainly, result in non-compliance and regulatory missteps.
So, if you think you can do everything by yourself, think again because a local representative can actually save your company money by:
- Making sure your product is launched on time,
- your company is saved from fines and penalties,
- there is no loss of licensing, market access denial, or product recalls.
Finding Expert cDMF Guidance
As we mentioned, having local expertise on your side is not only an advantage but a requirement to enter the Chinese pharmaceutical market. Partnering with experienced professionals can make a substantial difference in effectively managing these hurdles and streamlining the path to obtaining a Chinese Drug Master File (cDMF).
Partnering with Experts: Introducing Accestra Consulting
If you are looking for a local entity to help your company enter the Chinese market and need a cDMF, we would like to introduce Pharmaoffer’s new partner, Accestra. Accestra Consulting is an expert in Chinese regulatory affairs and offers specialized services that assist pharmaceutical companies in successfully navigating the cDMF process.
Accestra’s team of experts works in strategic locations like Hangzhou, Shanghai, and Beijing, leveraging local knowledge and connections with regulatory bodies to offer a smooth and positive pathway to your company’s market entry.
Case Study
Client: Company Y, a leading international API manufacturer, was looking to enter the Chinese market.
The company faced several issues related to its cDMF submission, particularly in the area of manufacturing process data. These were the main issues reported:
- Incomplete Production Process Flowchart and Reaction Equation:
Missing Key Details: The submitted flowchart and reaction equation were missing critical steps in the manufacturing process. Key details such as temperatures, pH levels, reaction times, and the sequence of reactions were not included. Additionally, crucial intermediates and by-products were not properly documented, leading to gaps in understanding the full process.
- Incomprehensive Process Description:
Lack of Clarity and Completeness: The process description failed to fully convey the manufacturing steps and controls in place. Specific areas of concern included:
Insufficient Detail on Critical Parameters: The description did not specify critical parameters such as temperature ranges, pressure settings, and control points for each step of the process.
Lack of In-Process Control Measures: Insufficient information was provided on the in-process control measures and quality checks conducted during manufacturing.
Missing Rationale for Process Steps: The rationale behind certain process steps and why specific conditions were chosen was not explained, leaving regulatory reviewers without a clear understanding of the process justification.
- Improper Selection of Starting Materials:
The starting materials were not properly selected, which did not meet the NMPA’s regulatory expectations. This included issues with the purity and source of the materials, as well as inadequate justification for their use in the manufacturing process.
Accestra Consulting was brought in to address these challenges. Here is how they provided comprehensive support to the client:
- Detailed Review and Revision of Flowcharts and Reaction Equations:
Accestra’s team worked closely with the client’s technical staff to ensure that the production process flowchart and reaction equation included all necessary details. This included specifying each process step, identifying key control points, and clearly outlining the reaction pathways.
- Enhancing Process Descriptions:
Assisted in revising the process description to ensure it was comprehensive and clear. They ensured that all steps were described in detail, including in-process controls and critical parameters that are crucial for regulatory compliance.
- Proper Selection and Justification of Starting Materials:
The team at Accestra guided the client in selecting appropriate starting materials and provided a detailed justification for their selection. This involved aligning the starting materials with regulatory expectations and providing thorough documentation to support their choice.
Outcome:
With Accestra’s expert guidance, Company Y successfully addressed the initial issues in their cDMF submission. The revised dossier met all regulatory requirements, significantly reducing the risk of delays or rejections.
Results:
Approval Success: The Company Y cDMF was approved by the NMPA without further queries, allowing them to proceed with their market entry plans.
Time Efficiency: The streamlined submission process saved the client significant time, ensuring a timely product launch.
Compliance Assurance: Company Y avoided potential fines, penalties, and market access denials, safeguarding its financial and operational interests. With a proven track record of over 100 successful product registrations, Accestra can offer tailored regulatory compliance and pharmacovigilance services that align with the NMPA regulations.
Find out more about by visiting Expert cDMF Guidance with Accestra
Conclusion
And there you have it— we hope you found everything you need to know about the Chinese Drug Master File (cDMF). Navigating China’s regulatory landscape might seem daunting, but this is what we do: break down complex themes to show you there is a way out of the labyrinth of information.
Whether you’re just starting or looking to update your processes, understanding and obtaining a cDMF will undoubtedly put you ahead of the game. Ready to make your mark in China? Well, roll up your sleeves and go get it.





